Research
Healthy humans are colonized by 500–1,000 different species of microbes, with effects on our physiology ranging from drug metabolism to cardiovascular health to immunomodulation. Even though we can now culture many of these microbes and sequence their genomes, however, we still lack insight into the function and regulation of their genes. This limits our ability to even form hypotheses about how these microbes affect host phenotypes.
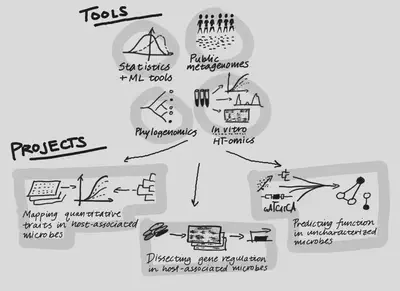
The Bradley Lab’s long-term aim is to understand our microbiota (especially the gut and oral microbiomes) as well as we currently understand model microbes, using both computational and high-throughput experimental tools. Such an understanding will allow us to interpret and eventually to modulate human microbiomes more precisely.
Current projects in the lab include:
-
Using natural variation to associate microbial genes with phenotypes. Prokaryotes of the human GI tract exhibit large variations in gene content, even within a single “species.” We have previously shown (Bradley et al., 2018; Bradley and Pollard, 2019) that we can use inter-species variation to discover microbial genes associated with prevalence at different body sites. Currently, we are developing methods to phenotype large numbers of isolates from the microbiome, and to use data about both inter- and intra-species variation to find microbial genes involved in specific aspects of host colonization.
-
Determining how gene regulation enables microbes to adapt to different body sites in health and disease. Changes in gene regulation are just as important as gene gain or loss in adaptation to an environment; however, regulatory changes are more difficult to detect and interpret. We will use comparative transcriptomics combined with new statistical methods to find potentially adaptive changes associated with microbial “lifestyle,” including associations with inflammatory diseases like Crohn’s.
-
Systematically predicting gene function in under-studied microbes. Integrating many high-throughput datasets has been shown in model systems to be a powerful method for generating hypotheses, not only about the biochemical functions of genes, but also the broader biological processes in which they participate (Bradley et al., 2019). We plan to use state-of-the-art machine learning tools together with omics-scale data to predict microbial gene functions directly, and to “close the loop” by testing these predictions in model systems.